ABSTRACT Carbon rings Cn and infinite chains C∞ are investigated by molecular-orbital and band-st... more ABSTRACT Carbon rings Cn and infinite chains C∞ are investigated by molecular-orbital and band-structure calculations within the local-density approximation. Carbon rings C4N (N<~8) exhibit a substantial first-order Jahn-Teller distortion that leads to long/short (single/triple) bond alternation decreasing with increasing N. Rings C4N+2 show no alternation (i.e., aromatic behavior is retained) until very large sizes (N>~20). For the infinite carbon chain uniform Brillouin-zone sampling with an even number of points Ns gives bond alternation. An odd number of sampling points gives no bond alternation for less than Ns=41. In the large Ns limit even and odd sampling lead to an upper and lower bound of 0.070a0 and 0.065a0 for bond alternation and 0.021–0.090 millihartrees/atom for condensation energy.
A new method of solution to the local spin density approximation to the electronic Schr\"{o}dinge... more A new method of solution to the local spin density approximation to the electronic Schr\"{o}dinger equation is presented. The method is based on an efficient, parallel, adaptive multigrid eigenvalue solver. It is shown that adaptivity is both necessary and sufficient to accurately solve the eigenvalue problem near the singularities at the atomic centers. While preliminary, these results suggest that direct real space methods may provide a much needed method for efficiently computing the forces in complex materials.
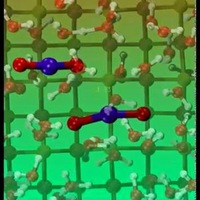
First principles molecular dynamics simulations of the hydration shells surrounding UO22+ ions ar... more First principles molecular dynamics simulations of the hydration shells surrounding UO22+ ions are reported for temperatures near 300 K. Most of the simulations were done with 64 solvating water molecules (22 ps). Simulations with 122 water molecules (9 ps) were also carried out. The hydration structure predicted from the simulations was found to agree with very well-known results from x-ray data. The average U=O bond length was found to be 1.77 A˚. The first hydration shell contained five trigonally coordinated water molecules that were equatorially oriented about the O-U-O axis with the hydrogen atoms oriented away from the uranium atom. The five waters in the first shell were located at an average distance of 2.44 A˚ (2.46 A˚, 122 water simulation). The second hydration shell was composed of distinct equatorial and apical regions resulting in a peak in the U-O radial distribution function at 4.59 A˚. The equatorial second shell contained ten water molecules hydrogen bonded to the five first shell molecules. Above and below the UO22+ ion, the water molecules were found to be significantly less structured. In these apical regions, water molecules were found to sporadically hydrogen bond to the oxygen atoms of the UO22+, oriented in such a way as to have their protons pointed toward the cation. While the number of apical waters varied greatly, an average of five to six waters was found in this region. Many water transfers into and out of the equatorial and apical second solvation shells were observed to occur on a picosecond time scale via dissociative mechanisms. Beyond these shells, the bonding pattern substantially returned to the tetrahedral structure of bulk water.
Results of the application of an adaptive finite element (FE) based solution using the FETK libra... more Results of the application of an adaptive finite element (FE) based solution using the FETK library of M. Holst to Density Functional Theory (DFT) approximation to the electronic structure of atoms and molecules are reported. The severe problem associated with the rapid variation of the electronic wave functions in the near singular regions of the atomic centers is treated by
Parallel hardware has become readily available to the computational chemistry research community.... more Parallel hardware has become readily available to the computational chemistry research community. This perspective will review the current state of parallel computational chemistry software utilizing high-performance parallel computing platforms. Hardware and software trends and their effect on quantum chemistry methodologies, algorithms, and software development will also be discussed.
The energies of isomers of C20 including neutral and positively charged ring, fused ring, flake, ... more The energies of isomers of C20 including neutral and positively charged ring, fused ring, flake, and fullerene structures have been calculated within the pseudopotential local density approximation (LDA). The objective is to predict the relative energies of the isomers as well as to validate LDA calculations for the carbon system. For C20, high-level coupled-cluster (CCSD (T)) calculations are just possible and are used for comparison. Our most accurate LDA calculations agree with prior calculations quantitatively and remarkably well with ...
This paper reports molecular dynamics simulations of the magnetite (001)-water interface, both in... more This paper reports molecular dynamics simulations of the magnetite (001)-water interface, both in pure water and in the presence of a 2.3 molal solution of NaClO4. The simulations are carried out using a potential model designed to allow the protonation states of the surface functional groups to evolve dynamically through the molecular dynamics trajectory. The primary structural quantities investigated are
ABSTRACT Carbon rings Cn and infinite chains C∞ are investigated by molecular-orbital and band-st... more ABSTRACT Carbon rings Cn and infinite chains C∞ are investigated by molecular-orbital and band-structure calculations within the local-density approximation. Carbon rings C4N (N<~8) exhibit a substantial first-order Jahn-Teller distortion that leads to long/short (single/triple) bond alternation decreasing with increasing N. Rings C4N+2 show no alternation (i.e., aromatic behavior is retained) until very large sizes (N>~20). For the infinite carbon chain uniform Brillouin-zone sampling with an even number of points Ns gives bond alternation. An odd number of sampling points gives no bond alternation for less than Ns=41. In the large Ns limit even and odd sampling lead to an upper and lower bound of 0.070a0 and 0.065a0 for bond alternation and 0.021–0.090 millihartrees/atom for condensation energy.
A new method of solution to the local spin density approximation to the electronic Schr\"{o}dinge... more A new method of solution to the local spin density approximation to the electronic Schr\"{o}dinger equation is presented. The method is based on an efficient, parallel, adaptive multigrid eigenvalue solver. It is shown that adaptivity is both necessary and sufficient to accurately solve the eigenvalue problem near the singularities at the atomic centers. While preliminary, these results suggest that direct real space methods may provide a much needed method for efficiently computing the forces in complex materials.
First principles molecular dynamics simulations of the hydration shells surrounding UO22+ ions ar... more First principles molecular dynamics simulations of the hydration shells surrounding UO22+ ions are reported for temperatures near 300 K. Most of the simulations were done with 64 solvating water molecules (22 ps). Simulations with 122 water molecules (9 ps) were also carried out. The hydration structure predicted from the simulations was found to agree with very well-known results from x-ray data. The average U=O bond length was found to be 1.77 A˚. The first hydration shell contained five trigonally coordinated water molecules that were equatorially oriented about the O-U-O axis with the hydrogen atoms oriented away from the uranium atom. The five waters in the first shell were located at an average distance of 2.44 A˚ (2.46 A˚, 122 water simulation). The second hydration shell was composed of distinct equatorial and apical regions resulting in a peak in the U-O radial distribution function at 4.59 A˚. The equatorial second shell contained ten water molecules hydrogen bonded to the five first shell molecules. Above and below the UO22+ ion, the water molecules were found to be significantly less structured. In these apical regions, water molecules were found to sporadically hydrogen bond to the oxygen atoms of the UO22+, oriented in such a way as to have their protons pointed toward the cation. While the number of apical waters varied greatly, an average of five to six waters was found in this region. Many water transfers into and out of the equatorial and apical second solvation shells were observed to occur on a picosecond time scale via dissociative mechanisms. Beyond these shells, the bonding pattern substantially returned to the tetrahedral structure of bulk water.
Results of the application of an adaptive finite element (FE) based solution using the FETK libra... more Results of the application of an adaptive finite element (FE) based solution using the FETK library of M. Holst to Density Functional Theory (DFT) approximation to the electronic structure of atoms and molecules are reported. The severe problem associated with the rapid variation of the electronic wave functions in the near singular regions of the atomic centers is treated by
Parallel hardware has become readily available to the computational chemistry research community.... more Parallel hardware has become readily available to the computational chemistry research community. This perspective will review the current state of parallel computational chemistry software utilizing high-performance parallel computing platforms. Hardware and software trends and their effect on quantum chemistry methodologies, algorithms, and software development will also be discussed.
The energies of isomers of C20 including neutral and positively charged ring, fused ring, flake, ... more The energies of isomers of C20 including neutral and positively charged ring, fused ring, flake, and fullerene structures have been calculated within the pseudopotential local density approximation (LDA). The objective is to predict the relative energies of the isomers as well as to validate LDA calculations for the carbon system. For C20, high-level coupled-cluster (CCSD (T)) calculations are just possible and are used for comparison. Our most accurate LDA calculations agree with prior calculations quantitatively and remarkably well with ...
This paper reports molecular dynamics simulations of the magnetite (001)-water interface, both in... more This paper reports molecular dynamics simulations of the magnetite (001)-water interface, both in pure water and in the presence of a 2.3 molal solution of NaClO4. The simulations are carried out using a potential model designed to allow the protonation states of the surface functional groups to evolve dynamically through the molecular dynamics trajectory. The primary structural quantities investigated are
Uploads
Papers by Eric Bylaska