As part of a recent trip to the University of Edinburgh to visit
Andrew Rambaut, I gave a talk on some work of mine published in the February edition of
Molecular Biology and Evolution and subsequently
recommended on the Faculty of 1000 website about the evolution of the gut pathogen
Campylobacter jejuni.
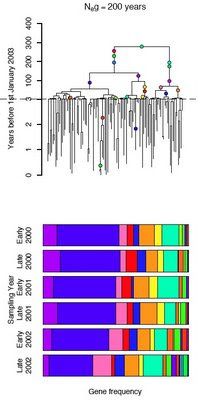
Part of the paper is concerned with the issue of the timescale of
Campylobacter evolution, and using longitudinal samples of
C. jejuni DNA sequences we attempted to calibrate the molecular clock in a similar way to that which is standard practice for viruses.
We detected surprisingly rapid evolution - 1,000 times faster than traditional estimates - which would place the split of
C. jejuni from its closest relative
C. coli during the Neolithic revolution. Interestingly, the point estimate of 6,500 years ago for the split from
C. coli - which preferentially infects swine - coincides with the
spread of pig domestication in the Near East and Europe in the 4th millennium BC.
The date is controversial because the
traditional dating method, which is based on bounding deep phylogenetic splits such as the common ancestor of mitochondria and bacteria, would place the divergence of
C. jejuni and
C. coli closer to 10 million years ago.
After the seminar I had an interesting discussion with
Paul Sharp, who was in the audience. Prof Sharp is actively researching the causes of conflict between long-term and short-term estimates of the rate of evolution in viruses. As he points out, short-term rate estimates (usually based on longitudinally-sampled viral sequences) frequently suggest that evolution is occurring much more rapidly than long-term estimates (based on deeper calibration points, such as co-phylogeny of host and pathogen). This phenomenon, observed in
HIV and
hepatitis C among others, may be caused by overly simplistic models of sequence evolution.
So how plausible is it that a ubiquitous bacterial pathogen such as
C. jejuni evolved as recently as the Neolithic, possibly in response to changes brought about by agriculture or animal husbandry? Longitudinal studies of
Helicobacter pylori and
Neisseria gonnorhoeae have obtained similarly rapid rates of bacterial evolution, and
evidence is mounting that the Neolithic revolution played an important role in creating new niches for human, plant and animal pathogens. Perhaps the best prospect for resolving these questions will be studies of ancient DNA preserved from the period in question.







 Posts
Posts





